
Disorders of the urea cycle
The urea cycle and potential defects
The urea cycle is the principal mechanism for clearing waste nitrogen resulting from protein turnover and for the metabolism of other nitrogenous metabolic compounds.1 It is also the only source of endogenous arginine, ornithine, and citrulline.1
The urea cycle includes 6 enzymes, 2 of which overlap with the nitric oxide pathway, as well as 2 amino acid transporters1:
- 5 catalytic enzymes
- Carbamylphosphate synthetase I (CPS1)
- Ornithine transcarbamylase (OTC)
- Argininosuccinic acid synthetase (ASS1)
- Argininosuccinic acid lyase (ASL)
- Arginase (ARG1)
- 1 cofactor-producing enzyme
- N-acetyl glutamate synthetase (NAGS)
- 2 amino acid transporters
- Ornithine translocase (ORNT1)
- Citrin
The urea cycle1*
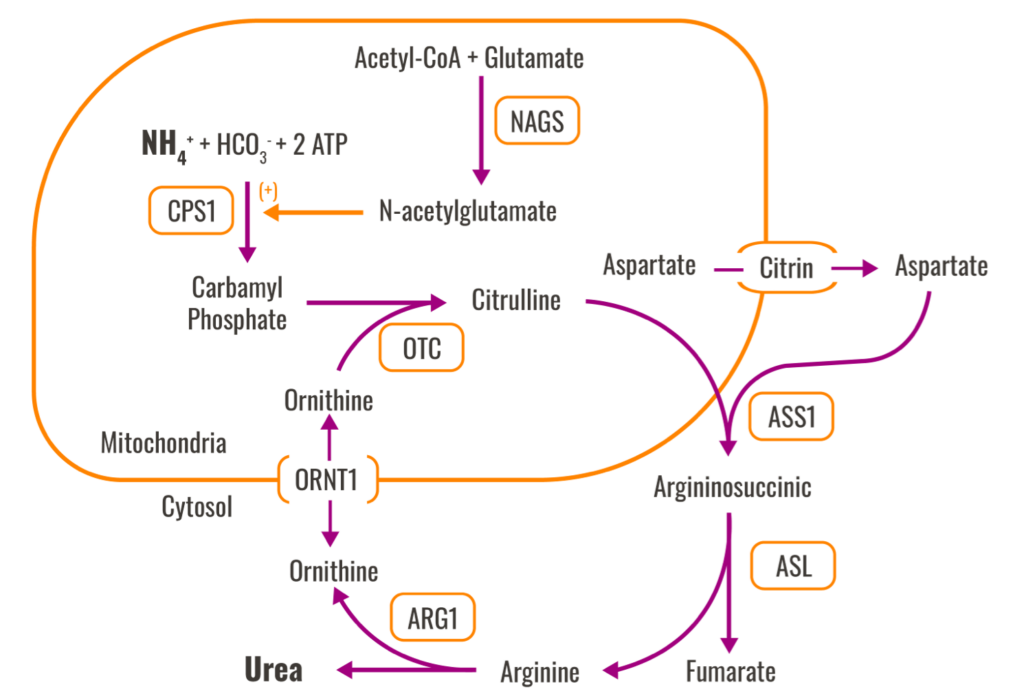
*This image has been adapted from its original source.1
Neonatal presentation of UCDs
Patients with severe UCDs usually present in the neonatal period.1 Such infants often appear normal at birth but rapidly develop cerebral edema and the related symptoms of lethargy, anorexia, hyper/hypoventilation, hypothermia, seizures, neurologic posturing, and coma.1
Typical early symptoms of hyperammonemia in children are nonspecific and include1,3,4 :
- Failure to feed
- Low core temperature
- Somnolence
Symptoms can then progress from somnolence to lethargy and coma.1
- Abnormal posturing and encephalopathy may occur1,3
- ~50% of neonates with severe hyperammonemia may have seizures, sometimes without overt clinical manifestations1
- The effects of hyperammonemia on the brain stem can cause hyperventilation that results in respiratory alkalosis1
- Hypoventilation and respiratory arrest follow as pressure on the brain stem increases1

UCD symptoms often develop at home, after a newborn has been discharged, and may not be readily recognized by the family or primary care physician.1

UCD symptoms often develop at home, after a newborn has been discharged, and may not be readily recognized by the family or primary care physician.1
Diagnosing UCDs
Diagnosis is based on clinical, biochemical, and molecular genetic data.1 A 3-generation family history should be obtained, with attention to other relatives with neurologic signs and symptoms of UCDs.1
Family history consistent with X-linked inheritance suggests OTC deficiency.1 Physical examination findings generally do not distinguish between the types of deficiencies.1
Some other disorders that affect the liver can also cause hyperammonemia, causing symptoms that mimic those of UCDs, so differential diagnosis is important.1 These include diseases of the liver and biliary tract, use of certain medications (eg, valproic acid, cyclophosphamide, 5-pentanoic acid), and certain genetic disorders.1
Molecular genetic testing is the definitive diagnostic test to confirm all types of UCDs.1

Molecular genetic testing is the definitive diagnostic test to confirm all types of UCDs.1


References
- Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea cycle disorders overview [updated June 22, 2017]. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews® [Internet]. University of Washington; 1993-2022. Accessed March 20, 2022. https://www.ncbi.nlm.nih.gov/books/NBK1217/
- McGuire PJ, Lee HS, members of the Urea Cycle Disorders Consortium, Summar ML. Infectious precipitants of acute hyperammonemia are associated with indicators of increased morbidity in patients with urea cycle disorders. J Pediatr. 2013;163(6):1705-1710.
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001;138(suppl 1):S30-S39.
- Kölker S, Garcia-Cazorla A, Valayannopoulos V, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015;38(6):1041-1057.
- Gardeitchik T, Humphrey M, Nation J, Boneh A. Early clinical manifestations and eating patterns in patients with urea cycle disorders. J Pediatr. 2012;161(2):328-332.